UCC Functions¶
Overview¶
In this notebook, we will introduce the basic functions of the UCC class. We will use UCCSD as a typical example. Other classes such as kUpCCGSD share the same interface.
We will show that, along with nice high-level interface, the UCC class also offers great flexibility and allows users to easily build their own algorithms.
Setup¶
[1]:
from tencirchem import UCCSD
from tencirchem.molecule import h2
TenCirChem provides a set of molecules in the molecule module for debugging and fast-prototyping.
TenCirChem uses PySCF Mole object to handle molecules. In other words, custom molecules can be built with exact the same interface as PySCF.
[2]:
h2
[2]:
<pyscf.gto.mole.Mole at 0x7f132bfd3990>
Hello world¶
This cell illustrates a simple UCC calculation.
[3]:
uccsd = UCCSD(h2)
uccsd.kernel()
uccsd.print_summary(include_circuit=True)
################################ Ansatz ###############################
#qubits #params #excitations initial condition
4 2 3 RHF
############################### Circuit ###############################
#qubits #gates #CNOT #multicontrol depth #FLOP
4 15 10 1 9 2160
############################### Energy ################################
energy (Hartree) error (mH) correlation energy (%)
HF -1.116706 20.568268 -0.000
MP2 -1.129868 7.406850 63.989
CCSD -1.137275 -0.000165 100.001
UCCSD -1.137274 0.000000 100.000
FCI -1.137274 0.000000 100.000
############################# Excitations #############################
excitation configuration parameter initial guess
0 (3, 2) 1001 1.082849e-16 0.000000
1 (1, 0) 0110 1.082849e-16 0.000000
2 (1, 3, 2, 0) 1010 -1.129866e-01 -0.072608
######################### Optimization Result #########################
e: -1.1372744055294384
fun: array(-1.13727441)
hess_inv: <2x2 LbfgsInvHessProduct with dtype=float64>
init_guess: [0.0, -0.07260814651571333]
jac: array([-9.60813938e-19, -1.11022302e-16])
message: 'CONVERGENCE: NORM_OF_PROJECTED_GRADIENT_<=_PGTOL'
nfev: 6
nit: 4
njev: 6
opt_time: 0.010929584503173828
staging_time: 3.337860107421875e-06
status: 0
success: True
x: array([ 1.08284918e-16, -1.12986561e-01])
UCC class has a convenient method print_summary for a quick glance of the calculation. The summary contains 5 parts:
Ansatz.
Circuit. TenCirChem by default uses a compact circuit for excitations (see the Circuit section below), in which multi-control gates are involved. The circuit part also gives an estimation of the FLOP required for a full statevector simulation of the circuit.
Energy. Includes optimized UCC energy along with HF, MP2, CCSD and FCI energy for reference.
Excitations. Includes optimized UCC parameters and their initial guess. See the documentation for the convention of excitation operators and configurations.
Optimization Results. This is simply a modified SciPy
OptimizeResultobject.
print_summary by default will not print circuit information since it is sometimes significantly more time-consuming than other parts.
One may print different parts of the summary separately using corresponding standalone method such as print_ansatz.
The Circuit¶
TenCirChem uses TensorCircuit circuit object for quantum circuit.
[4]:
circuit = uccsd.get_circuit()
circuit
[4]:
<tensorcircuit.circuit.Circuit at 0x7f131c17fe10>
[5]:
circuit.draw(output="mpl")
[5]:
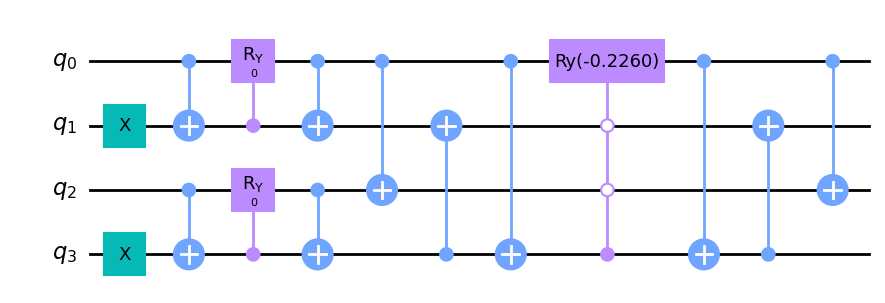
TenCirChem uses the compact circuit for excitations described in https://arxiv.org/pdf/2005.14475.pdf. To decompose the multicontroled \(R_y\) gate into elementary gates, use the decompose_multicontrol argument.
[6]:
uccsd.get_circuit(decompose_multicontrol=True).draw(output="mpl")
[6]:
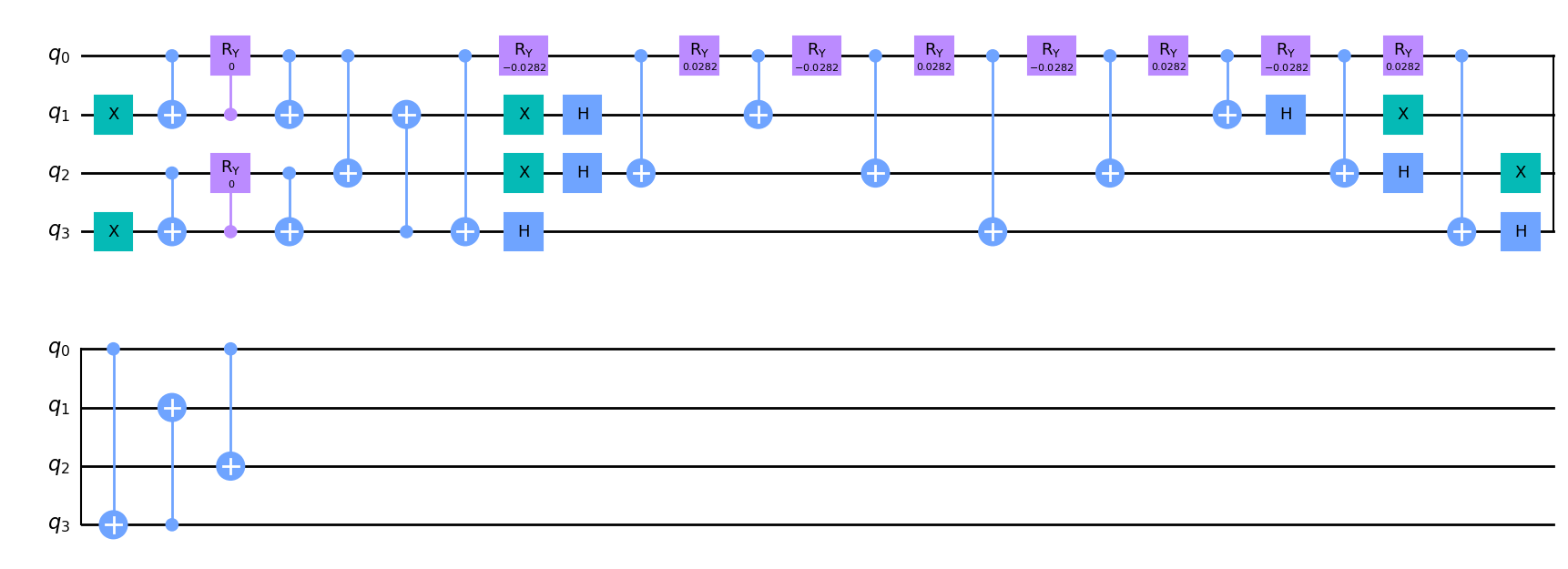
For traditional Trotterized circuit, use the trotter argument.
[7]:
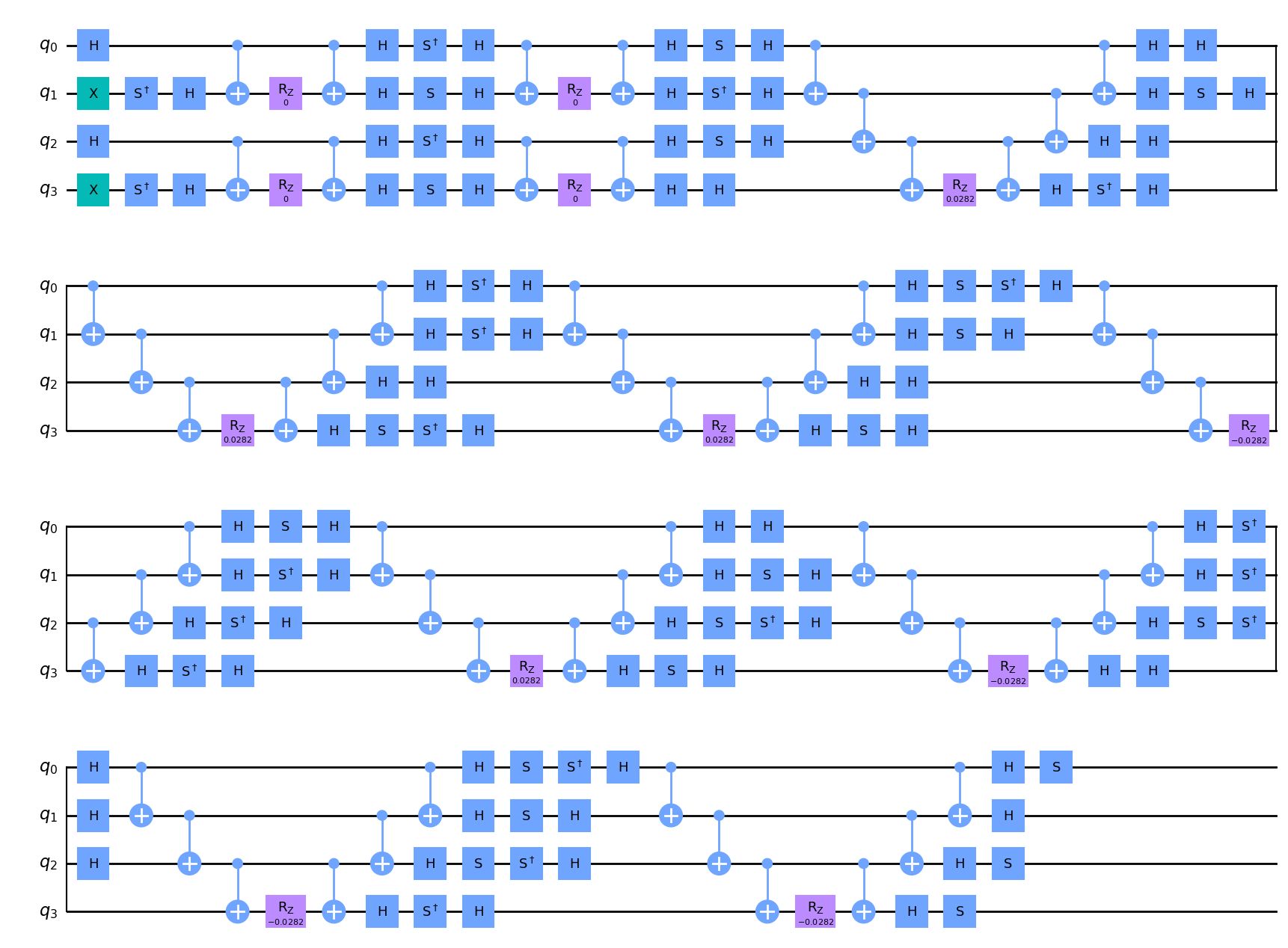
uccsd.get_circuit(trotter=True).draw(output="mpl")
[7]:
Useful Attributes¶
TenCirChem chooses to expose its internal data whenever possible and in the simplest way. The intention is to make TenCirChem easy to hack and useful as a handy toolbox.
[8]:
uccsd.n_qubits, uccsd.n_elec
[8]:
(4, 2)
[9]:
# PySCF objects
uccsd.mol, uccsd.hf
[9]:
(<pyscf.gto.mole.Mole at 0x7f13232ed950>,
<pyscf.scf.hf_symm.SymAdaptedRHF at 0x7f132bafc850>)
[10]:
uccsd.e_hf, uccsd.e_mp2, uccsd.e_ccsd, uccsd.e_ucc, uccsd.e_fci, uccsd.e_nuc
[10]:
(-1.116706137236105,
-1.1298675557838804,
-1.1372745709766439,
-1.1372744055294384,
-1.1372744055294384,
0.7141392859919029)
[11]:
# one and two electron integral in molecular orbital basis
uccsd.int1e.shape, uccsd.int2e.shape
[11]:
((2, 2), (2, 2, 2, 2))
[12]:
# Hamiltonian as openfermion FermionOperator
uccsd.h_fermion_op
[12]:
0.7141392859919029 [] +
-1.2527052599711868 [0^ 0] +
-0.48227117798977825 [0^ 1^ 0 1] +
-0.6745650967143663 [0^ 2^ 0 2] +
-0.18126641677772592 [0^ 2^ 1 3] +
-0.6635375947675042 [0^ 3^ 0 3] +
-0.18126641677772592 [0^ 3^ 1 2] +
-0.47569770336145906 [1^ 1] +
-0.18126641677772592 [1^ 2^ 0 3] +
-0.6635375947675038 [1^ 2^ 1 2] +
-0.18126641677772592 [1^ 3^ 0 2] +
-0.6974673850129379 [1^ 3^ 1 3] +
-1.2527052599711868 [2^ 2] +
-0.48227117798977825 [2^ 3^ 2 3] +
-0.47569770336145906 [3^ 3]
[13]:
# Hamiltonian as openfermion QubitOperator
uccsd.h_qubit_op
[13]:
(-0.09835117053027564+0j) [] +
(0.04531660419443148+0j) [X0 X1 X2 X3] +
(0.04531660419443148+0j) [X0 X1 Y2 Y3] +
(0.04531660419443148+0j) [Y0 Y1 X2 X3] +
(0.04531660419443148+0j) [Y0 Y1 Y2 Y3] +
(-0.22297018776182556+0j) [Z0] +
(0.12056779449744456+0j) [Z0 Z1] +
(0.17436684625323448+0j) [Z0 Z2] +
(0.16588439869187604+0j) [Z0 Z3] +
(0.1712591626176813+0j) [Z1] +
(0.16588439869187596+0j) [Z1 Z2] +
(0.16864127417859157+0j) [Z1 Z3] +
(-0.22297018776182548+0j) [Z2] +
(0.12056779449744456+0j) [Z2 Z3] +
(0.1712591626176812+0j) [Z3]
[14]:
# t1, t2 amplitude based on MP2 or CCSD (determined by the `init_method` argument)
uccsd.t1, uccsd.t2
[14]:
(None, array([[[[-0.07260815]]]]))
[15]:
# `param_ids` maps excitation operators to parameters.
# Some excitation operators share the same parameter due to symmetry
# refer to the documentation for the convention of excitation operators
uccsd.ex_ops, uccsd.param_ids, uccsd.init_guess
[15]:
([(3, 2), (1, 0), (1, 3, 2, 0)], [0, 0, 1], [0.0, -0.07260814651571333])
[16]:
# Scipy `OptimizeResult` object
uccsd.opt_res
[16]:
e: -1.1372744055294384
fun: array(-1.13727441)
hess_inv: <2x2 LbfgsInvHessProduct with dtype=float64>
init_guess: [0.0, -0.07260814651571333]
jac: array([-9.60813938e-19, -1.11022302e-16])
message: 'CONVERGENCE: NORM_OF_PROJECTED_GRADIENT_<=_PGTOL'
nfev: 6
nit: 4
njev: 6
opt_time: 0.010929584503173828
staging_time: 3.337860107421875e-06
status: 0
success: True
x: array([ 1.08284918e-16, -1.12986561e-01])
[17]:
# the optimized parameters
uccsd.params
[17]:
array([ 1.08284918e-16, -1.12986561e-01])
[18]:
# optimized circuit statevector
uccsd.statevector()
[18]:
array([ 0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00, 9.93623806e-01, 1.08284918e-16, 0.00000000e+00,
0.00000000e+00, 1.08284918e-16, -1.12746318e-01, 0.00000000e+00,
0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00])
[19]:
# configuration interaction vector
uccsd.civector()
[19]:
array([ 9.93623806e-01, 1.08284918e-16, 1.08284918e-16, -1.12746318e-01])
[20]:
# bitstring for each element in the CI vector
uccsd.get_ci_strings()
[20]:
array([ 5, 6, 9, 10], dtype=uint64)
Active Space Approximation¶
TenCirChem offers the simplest API for active space approximation among available packages.
[21]:
from tencirchem.molecule import h8
# (2e, 2o) active space
uccsd = UCCSD(h8, active_space=(2, 2))
uccsd.kernel()
uccsd.print_summary()
################################ Ansatz ###############################
#qubits #params #excitations initial condition
4 2 3 RHF
############################### Energy ################################
energy (Hartree) error (mH) correlation energy (%)
HF -4.149619 1.343542e+01 -0.000
MP2 -4.157573 5.480558e+00 59.208
CCSD -4.163054 -1.932076e-04 100.001
UCCSD -4.163054 -8.881784e-13 100.000
FCI -4.163054 0.000000e+00 100.000
############################# Excitations #############################
excitation configuration parameter initial guess
0 (3, 2) 1001 1.112312e-15 0.000000
1 (1, 0) 0110 1.112312e-15 0.000000
2 (1, 3, 2, 0) 1010 -1.377274e-01 -0.082065
######################### Optimization Result #########################
e: -4.163053957291894
fun: array(-4.16305396)
hess_inv: <2x2 LbfgsInvHessProduct with dtype=float64>
init_guess: [0.0, -0.08206541779079227]
jac: array([-1.21074365e-17, -5.55111512e-17])
message: 'CONVERGENCE: NORM_OF_PROJECTED_GRADIENT_<=_PGTOL'
nfev: 6
nit: 4
njev: 6
opt_time: 0.003999471664428711
staging_time: 1.6689300537109375e-06
status: 0
success: True
x: array([ 1.11231160e-15, -1.37727434e-01])
The reference energies including MP2, CCSD nad FCI are also based on the active space.
Starting from the Integrals¶
One may start the UCC calculation from custom integrals without the need to define a molecule.
[22]:
uccsd_from_integral = UCCSD.from_integral(uccsd.int1e, uccsd.int2e, uccsd.n_elec, uccsd.e_core)
# the energy is the same with the last calculation
print(uccsd_from_integral.kernel(), uccsd.e_ucc)
-4.163053957291894 -4.163053957291894
Feeding in Custom Parameters¶
In TenCirChem, it is also fairly easy to feed in customized parameters to get desired properties
[23]:
import numpy as np
params = np.zeros(uccsd.n_params)
[24]:
uccsd.energy(params), uccsd.e_hf
[24]:
(-4.149618533807672, -4.1496185338076685)
[25]:
# energy and gradient, can be used in optimization
# one caveat is that TenCirChem by default uses float32, while SciPy assumes float64
uccsd.energy_and_grad(params)
[25]:
(-4.149618533807672, array([-2.27679890e-15, 1.93866451e-01]))
[26]:
uccsd.statevector(params)
[26]:
array([0., 0., 0., 0., 0., 1., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0.])
[27]:
# Configuration Interaction vector. Compatible with PySCF.
civector = uccsd.civector(params)
civector
[27]:
array([1., 0., 0., 0.])
[28]:
# calculate the energy by hand
civector @ uccsd.hamiltonian(civector) + uccsd.e_core
[28]:
-4.149618533807672
[29]:
# set the parameters, and use the class as usual
uccsd.params = params
uccsd.print_summary()
################################ Ansatz ###############################
#qubits #params #excitations initial condition
4 2 3 RHF
############################### Energy ################################
energy (Hartree) error (mH) correlation energy (%)
HF -4.149619 13.435423 -0.000
MP2 -4.157573 5.480558 59.208
CCSD -4.163054 -0.000193 100.001
UCCSD -4.149619 13.435423 0.000
FCI -4.163054 0.000000 100.000
############################# Excitations #############################
excitation configuration parameter initial guess
0 (3, 2) 1001 0.0 0.000000
1 (1, 0) 0110 0.0 0.000000
2 (1, 3, 2, 0) 1010 0.0 -0.082065
######################### Optimization Result #########################
e: -4.163053957291894
fun: array(-4.16305396)
hess_inv: <2x2 LbfgsInvHessProduct with dtype=float64>
init_guess: [0.0, -0.08206541779079227]
jac: array([-1.21074365e-17, -5.55111512e-17])
message: 'CONVERGENCE: NORM_OF_PROJECTED_GRADIENT_<=_PGTOL'
nfev: 6
nit: 4
njev: 6
opt_time: 0.003999471664428711
staging_time: 1.6689300537109375e-06
status: 0
success: True
x: array([ 1.11231160e-15, -1.37727434e-01])
Reduced Density Matrix¶
Calculate reduced density matrix in atomic orbital or molecule orbital basis
[30]:
uccsd.make_rdm1().shape, uccsd.make_rdm2().shape
[30]:
((8, 8), (8, 8, 8, 8))
[31]:
uccsd.make_rdm1(basis="MO").shape, uccsd.make_rdm2(basis="MO").shape
[31]:
((8, 8), (8, 8, 8, 8))
Note that the RDM is for the whole space, beyond the active space.